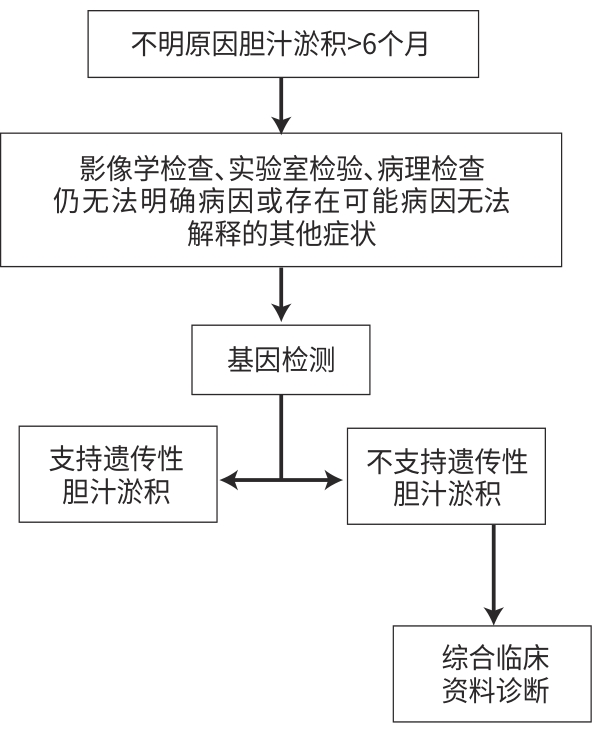
| [1] |
Chinese Society of Hepatology, Chinese Medical Association. Guidelines on the management of cholestasis liver diseases(2021)[J]. J Clin Hepatol, 2022, 38( 1): 62- 69. DOI: 10.3969/j.issn.1001-5256.2022.01.010. |
| [2] |
BORTOLINI M, ALMASIO P, BRAY G, et al. Multicentre survey of the prevalence of intrahepatic cholestasis in 2520 consecutive patients with newly diagnosed chronic liver disease[J]. Drug Investig, 1992, 4( 4): 83- 89. DOI: 10.1007/BF03258368. |
| [3] |
NG SB, BUCKINGHAM KJ, LEE C, et al. Exome sequencing identifies the cause of a Mendelian disorder[J]. Nat Genet, 2010, 42( 1): 30- 35. DOI: 10.1038/ng.499. |
| [4] |
BOTSTEIN D, RISCH N. Discovering genotypes underlying human phenotypes: Past successes for Mendelian disease, future approaches for complex disease[J]. Nat Genet, 2003, 33 Suppl: 228- 237. DOI: 10.1038/ng1090. |
| [5] |
European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Management of cholestatic liver diseases[J]. J Hepatol, 2009, 51( 2): 237- 267. DOI: 10.1016/j.jhep.2009.04.009. |
| [6] |
GUNAYDIN M, BOZKURTER CIL AT. Progressive familial intrahepatic cholestasis: Diagnosis, management, and treatment[J]. Hepat Med, 2018, 10: 95- 104. DOI: 10.2147/HMER.S137209. |
| [7] |
GUO J, XIE SY, YANG LR, et al. Clinical characteristics of 2 cases of progressive familial intrahepatic cholestasis and literature review[J/CD]. Chin J Liver Dis(Electronic Version), 2024, 16( 1): 67- 72. DOI: 10.3969/j.issn.1674-7380.2024.01.012. |
| [8] |
WENG YH, XIONG QF, LIU DX, et al. Clinical and pathological features of progressive familial intrahepatic cholestasis type 3[J]. J Clin Hepatol, 2022, 38( 1): 154- 159. DOI: 10.3969/j.issn.1001-5256.2022.01.024. |
| [9] |
GOTTHARDT D, RUNZ H, KEITEL V, et al. A mutation in the canalicular phospholipid transporter gene, ABCB4 is associated with cholestasis, ductopenia, and cirrhosis in adults[J]. Hepatology, 2008, 48( 4): 1157- 1166. DOI: 10.1002/hep.22485. |
| [10] |
HALAWI A, IBRAHIM N, BITAR R. Triggers of benign recurrent intrahepatic cholestasis and its pathophysiology: A review of literature[J]. Acta Gastroenterol Belg, 2021, 84( 3): 477- 486. DOI: 10.51821/84.3.013. |
| [11] |
CHEN FF, PENG LQ, WANG J. Correlation between serum glycocholic acid and liver biochemical indexes and inflammatory cytokines in patients with intrahepatic chole-stasis of pregnancy[J]. J Clin Exp Med, 2023, 22( 14): 1534- 1537. DOI: 10.3969/j.issn.1671-4695.2023.14.021. |
| [12] |
Obstetrics Subgroup, Society of Obstetrics and Gynecology, Chinese Medical Association; Society of Perinatal Medicine, Chinese Medical Association. Guidelines for clinical diagnosis, treatment and management of intrahepatic cholestasis of pregnancy(2024)[J]. Chin J Obstet Gynecol, 2024, 59( 2): 97- 107. DOI: 10.3760/cma.j.cn112141-20230914-00099. |
| [13] |
AYDıN GA, ÖZGEN G, GÖRÜKMEZ O. The role of genetic mutations in intrahepatic cholestasis of pregnancy[J]. Taiwan J Obstet Gynecol, 2020, 59( 5): 706- 710. DOI: 10.1016/j.tjog.2020.07.014. |
| [14] |
NAYAGAM JS, WILLIAMSON C, JOSHI D, et al. Review article: Liver disease in adults with variants in the cholestasis-related genes ABCB11 ABCB4 and ATP8B1[J]. Aliment Pharmacol Ther, 2020, 52( 11-12): 1628- 1639. DOI: 10.1111/apt.16118. |
| [15] |
AYOUB MD, KAMATH BM. Alagille syndrome: Current understanding of pathogenesis, and challenges in diagnosis and management[J]. Clin Liver Dis, 2022, 26( 3): 355- 370. DOI: 10.1016/j.cld.2022.03.002. |
| [16] |
LI LT, DONG JB, WANG XH, et al. JAG1 mutation spectrum and origin in Chinese children with clinical features of alagille syndrome[J]. PLoS One, 2015, 10( 6): e0130355. DOI: 10.1371/journal.pone.0130355. |
| [17] |
Inherited Metabolic Liver Disease Collaboration Group, Chinese Society of Hepatology, Chinese Medical Association. Guidelines for the diagnosis and treatment of hepatolenticular degeneration(2022 edition)[J]. Chin J Hepatol, 2022, 30( 1): 9- 20. DOI: 10.3760/cma.j.cn501113-20211217-00603. |
| [18] |
DANKS DM. Copper and liver disease[J]. Eur J Pediatr, 1991, 150( 3): 142- 148. DOI: 10.1007/BF01963553. |
| [19] |
HASBAOUI BE, RIFAI Z, SAGHIR S, et al. Congenital hepatic fibrosis: Case report and review of literature[J]. Pan Afr Med J, 2021, 38: 188. DOI: 10.11604/pamj.2021.38.188.27941. |
| [20] |
Writing Group For Practice Guidelines For Diagnosis And Treatment Of Genetic Diseases Medical Genetics Branch Of Chinese Medical Association. Clinical practice guidelines for polycystic kidney diseases[J]. Chin J Med Genet, 2020, 37( 3): 277- 283. DOI: 10.3760/cma.j.issn.1003-9406.2020.03.009. |
| [21] |
|
| [22] |
LANKTREE MB, HAGHIGHI A, di BARI I, et al. Insights into autosomal dominant polycystic kidney disease from genetic studies[J]. Clin J Am Soc Nephrol, 2021, 16( 5): 790- 799. DOI: 10.2215/CJN.02320220. |
| [23] |
DIXIT A, PATEL C, HARRISON R, et al. 17q12 microdeletion syndrome: Three patients illustrating the phenotypic spectrum[J]. Am J Med Genet A, 2012, 158 A( 9): 2317- 2321. DOI: 10.1002/ajmg.a.35520. |
| [24] |
Technology Committee on DILI Prevention and Management, Chinese Medical Biotechnology Association; Study Group of Drug-Induced Liver Disease, Chinese Medical Association for the Study of Liver Diseases. Chinese guideline for diagnosis and management of drug-induced liver injury(2023 version)[J]. Chin J Gastroenterol, 2023, 28( 7): 397- 431. DOI: 10.3760/cma.j.cn501113-20230419-00176. |
| [25] |
Biliary Surgery Group of the Surgery Branch of the Chinese Medical Association. Guidelines for diagnosis and treatment of hepatolithiasis[J]. Chin J Dig Surg, 2007, 6( 2): 156- 161. DOI: 10.3760/cma.j.issn.1673-9752.2007.02.028. |
| [26] |
YAO X, LIU Y, YU LD, et al. Rare portal hypertension caused by abernethy malformation(type IIC): A case report[J]. World J Radiol, 2023, 15( 8): 250- 255. DOI: 10.4329/wjr.v15.i8.250. |
| [27] |
ENNAIFER R, BACHA D, ROMDHANE H, et al. Budd-chiari syndrome: An unusual presentation of multisystemic sarcoidosis[J]. Clin Pract, 2015, 5( 3): 768. DOI: 10.4081/cp.2015.768. |
| [28] |
WOJCIK MH, LEMIRE G, BERGER E, et al. Genome sequencing for diagnosing rare diseases[J]. N Engl J Med, 2024, 390( 21): 1985- 1997. DOI: 10.1056/NEJMoa2314761. |






 DownLoad:
DownLoad: