
 PDF下载 ( 2080 KB)
PDF下载 ( 2080 KB)

26例先天性肝纤维化患者的临床及病理特点分析
DOI: 10.12449/JCH251118
Clinical and pathological features of patients with congenital hepatic fibrosis: An analysis of 26 cases
-
摘要:
目的 总结先天性肝纤维化(CHF)患者的临床特征及病理学特点,并分析比较不同年龄段患者的临床及病理特征差异。 方法 回顾性分析2005年8月—2023年6月在中日友好医院经病理学确诊为CHF的26例患者的临床病理资料;将这些患者进行年龄分层,分析不同年龄段患者的临床及病理学特征。 结果 26例患者中男12例,女14例,发病年龄在4~61岁;门静脉高压型19例(73.08%),胆管炎型2例(7.69%),混合型4例(15.38%),隐匿型1例(3.85%);除4例临床症状不详外,近半数患者因门静脉高压所致的上消化道出血(10/22,45.45%)就诊。病理学表现均可见汇管区宽阔的纤维间隔分隔大致正常的肝实质,伴有异常的胆管反应性增生,在4例10岁前起病的儿童患者中可见更为致密的纤维间隔、缺乏相应口径的门静脉,且代偿性的薄壁血管显著减少,甚至消失。 结论 CHF临床上以门静脉高压型最为常见,早期起病患者的肝组织病理学具有一定特点,并可能与早期出现门静脉高压相关的严重并发症相关,因此对临床可疑的患者进行肝穿刺,必要时进行基因检测,早发现、早诊断,对改善患者预后尤为重要。 -
关键词:
- 先天性肝纤维化 /
- 病理状态, 体征和症状 /
- 门静脉高压
Abstract:Objectives To summarize the clinical and pathological features of patients with congenital liver fibrosis (CHF), and to investigate the differences in clinical and pathological features between patients in different age groups. Methods A retrospective analysis was performed for the clinicopathological data of 26 patients with pathologically confirmed CHF in China-Japan Friendship Hospital from August 2005 to June 2023, and the patients were stratified by age to investigate the clinical and pathological features of patients in different age groups. Results Among the 26 patients, there were 12 male patients and 14 female patients, with an age of onset of 4 — 61 years. There were 19 patients with portal hypertension type (73.08%), 2 patients with cholangitis type (7.69%), 4 patients with mixed type (15.38%), and 1 patient with occult type (3.85%). Of all 26 patients, 4 had unknown clinical symptoms, and among the 22 patients with clear clinical symptoms, 10 (45.45%) attended the hospital due to upper gastrointestinal bleeding caused by portal hypertension. Pathological manifestations included roughly normal liver parenchyma separated by fibrous septa in the portal area, with the presence of abnormal reactive bile duct hyperplasia. Denser fibrous septa and a lack of portal veins with the corresponding caliber were observed in 4 pediatric patients with disease onset before the age of 10 years, with a significant reduction or even disappearance of compensatory thin-walled blood vessels. Conclusion Portal hypertension-type CHF is the most common type in clinical practice. Patients with an early age of onset have certain histopathological features of the liver, with the presence of serious complications associated with portal hypertension. Therefore, liver biopsy should be performed for patients suspected of CHF in clinical practice, and genetic testing should be performed when necessary. Early identification and diagnosis are of great importance for improving the prognosis of patients. -

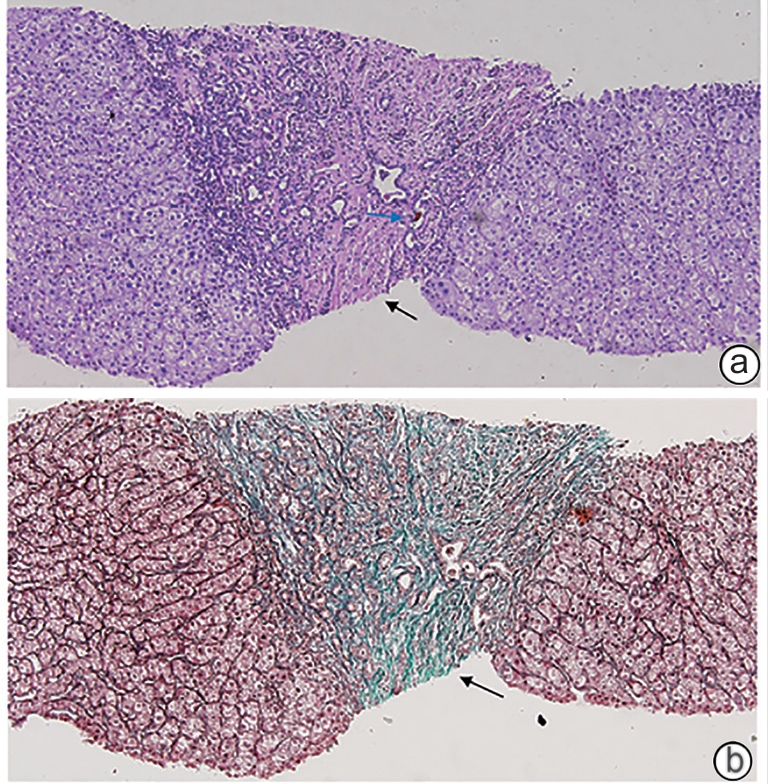
注: 男性,23岁。a(HE染色,×40)、b(HE染色,×100):肝实质为宽阔的纤维间隔不规则分隔(黑色箭头示纤维间隔),间隔为纤维性,其内可见增生的、多数形状不规则的小胆管,少数呈分支状,有的轻度扩张含浓缩胆汁,间隔内可见小动脉,未见相应口径的门静脉;间隔与肝实质界清,部分交界带见细胆管反应性增生。c(CK7免疫组化染色,×100):显示交界带内增生的细胆管。d(网织+Masson染色,×100):显示宽阔的纤维间隔(黑色箭头示纤维间隔)。
图 1 CHF的病理学改变
Figure 1. Pathological changes in CHF
注: 男性,24岁,伴Caroli病。a(HE染色,×100):肝实质为多条宽阔纤维带所分隔(黑色箭头示纤维间隔区域),纤维带内有的可见多数大小不一的幼稚小胆管,少数管腔内含胆汁(蓝色箭头示胆管内淤积的胆汁);有的含扩张的胆管,衬附单层立方上皮或矮柱状上皮,周围未见明显炎症。肝实质内肝板排列尚整,部分肝细胞内含脂褐素,偶见小坏死灶。b(网织+Masson染色,×100):显示宽阔的纤维间隔区域(黑色箭头示纤维间隔区域)。
图 2 CHF合并Caroli病的病理学改变
Figure 2. Pathological changes of CHF with Caroli disease
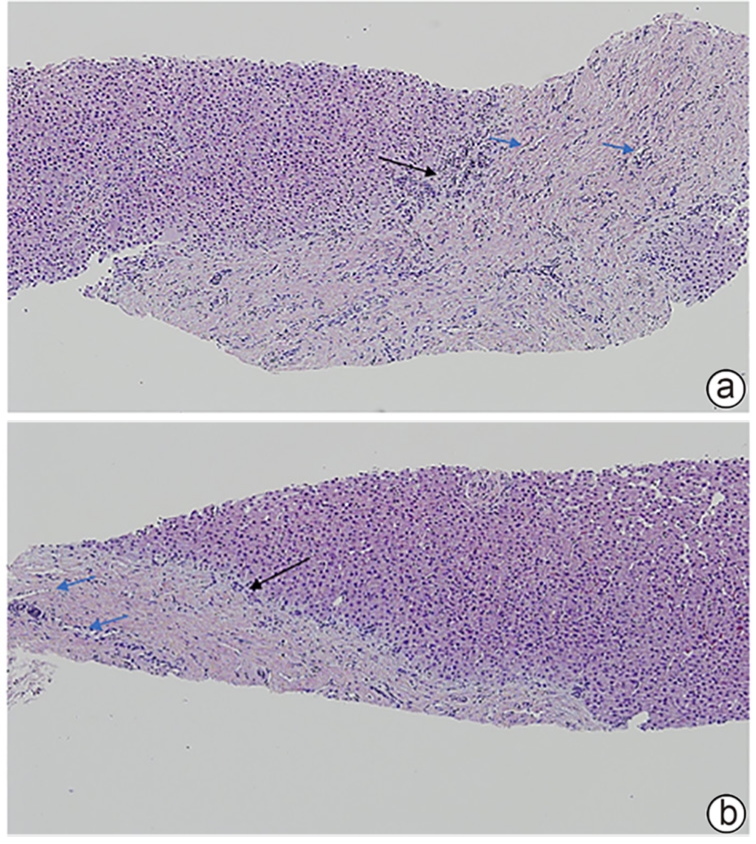
注: 女性,4岁。a、b(HE染色,×100):肝穿刺组织内见宽阔而边界整齐的纤维间隔(黑色箭头示纤维间隔)。间隔内纤维玻变、胶原致密,炎症轻,门静脉消失,仅有个别裂隙样的薄壁小血管(蓝色箭头表示薄壁小血管)。
图 3 儿童CHF患者的病理学改变
Figure 3. Pathological changes of CHF in pediatric patients
表 1 不同年龄段患者实验室检验结果
Table 1. Laboratory test results in patients of different age groups
指标 儿童组(n=4) 少年组(n=4) 青壮年组(n=11) 中老年组(n=7) ALT(U/L) 65.00(47.05~261.00) 55.50(21.00~187.50) 13.00(8.50~29.50) 17.00(8.18~27.33) AST(U/L) 55.00(27.50~172.50) 62.50(5.50~145.75) 19.00(15.00~29.50) 22.05(17.88~30.75) ALP(U/L) 206.60(103.30~403.80) 194.50(62.50~373.00) 79.00(57.00~203.00) 121.50(87.25~187.30) GGT(U/L) 89.00(64.85~193.50) 90.00(28.00~290.00) 37.00(19.00~287.50) 103.00(46.00~163.05) TBil(μmol/L) 15.10±0.60 41.36±21.53 30.44±41.30 18.31±7.62 DBil(μmol/L) 6.70±0.50 19.02±12.84 20.91±41.80 6.02±3.21 Alb(g/L) 32.10±4.10 31.02±17.11 36.50±6.49 37.37±6.48 GLO(g/L) 26.60±2.00 18.83±16.38 27.58±8.47 33.36±4.31 TBA(μmol/L) 6.30±0.60 7.30±0.20 17.11±19.62 17.42±13.07 WBC(×109/L) 3.35±1.35 3.30±1.50 4.59±3.59 4.96±2.27 RBC(×109/L) 3.43±0.90 3.31±0.20 3.41±0.84 3.25±0.96 Hb(g/L) 101.00±2.10 62.00±2.40 93.67±31.31 95.86±28.76 PLT(×109/L) 129.50±53.50 89.00±3.10 175.93±214.55 149.00±29.93 注:GLO,球蛋白;TBA,总胆汁酸;WBC,白细胞计数;RBC,红细胞计数;Hb,血红蛋白。儿童组有2例患者TBil、DBil、GLO水平及血常规结果缺失,少年组有2例患者血常规结果缺失,青壮年组及中老年组各有1例患者血常规结果缺失。
 下载: 导出CSV
下载: 导出CSV
表 2 不同年龄段患者的临床症状
Table 2. Clinical symptoms in patients of different age groups
症状 儿童组 少年组 青壮年组 中老年组 呕血、黑便 3/4(75.00%) 2/4(50.00%) 5/10(50.00%) 0/4(0.00%) 黄疸 1/4(25.00%) 1/4(25.00%) 4/10(40.00%) 1/4(25.00%) 腹痛、腹胀 0/4(0.00%) 1/4(25.00%) 4/10(40.00%) 0/4(0.00%) 肝/脾肿大 3/4(75.00%) 2/4(50.00%) 9/10(90.00%) 4/4(100.00%) 发热 0/4(0.00%) 0/4(0.00%) 2/10(20.00%) 1/4(25.00%) 肝功能异常 3/4(75.00%) 2/4(50.00%) 4/11(36.36%) 6/7(85.71%) 食管胃底静脉曲张 2/2(100.00%) 3/4(75.00%) 5/6(83.33%) 2/3(66.67%) 注:儿童组中仅2例进行影像学检查;青壮年组中10例汇报临床症状,其中6例进行影像学检查;中老年组中4例汇报临床症状,3例进行影像学检查。
下载: 导出CSV
-
[1] LASAGNI A, CADAMURO M, MORANA G, et al. Fibrocystic liver disease: Novel concepts and translational perspectives[J]. Transl Gastroenterol Hepatol, 2021, 6: 26. DOI: 10.21037/tgh-2020-04. [2] WU X, ZHOU C, LUO SQ. Congenital hepatic fibrosis: Clinical features of different clinical types in 75 patients[J]. Chin Hepatol, 2014, 19( 7): 479- 482, 490. DOI: 10.14000/j.cnki.issn.1008-1704.2014.07.002.吴欣, 周超, 罗生强. 先天性肝纤维化不同分型的临床特征: 75例分析[J]. 肝脏, 2014, 19( 7): 479- 482, 490. DOI: 10.14000/j.cnki.issn.1008-1704.2014.07.002. [3] CHEN IY, WHITNEY-MILLER CL, LIAO XY. Congenital hepatic fibrosis and its mimics: A clinicopathologic study of 19 cases at a single institution[J]. Diagn Pathol, 2021, 16( 1): 81. DOI: 10.1186/s13000-021-01142-y. [4] ALSOMALI MI, YEARSLEY MM, LEVIN DM, et al. Diagnosis of congenital hepatic fibrosis in adulthood[J]. Am J Clin Pathol, 2020, 153( 1): 119- 125. DOI: 10.1093/ajcp/aqz140. [5] JIANG L, FANG PP, WEEMHOFF JL, et al. Evidence for a“pathogenic triumvirate” in congenital hepatic fibrosis in autosomal recessive polycystic kidney disease[J]. BioMed Res Int, 2016, 2016: 4918798. DOI: 10.1155/2016/4918798. [6] DORVAL G, BOYER O, COUDERC A, et al. Long-term kidney and liver outcome in 50 children with autosomal recessive polycystic kidney disease[J]. Pediatr Nephrol, 2021, 36( 5): 1165- 1173. DOI: 10.1007/s00467-020-04808-9. [7] LOOMES KM, RUSSO P, RYAN M, et al. Bile duct proliferation in liver-specific Jag1 conditional knockout mice: Effects of gene dosage[J]. Hepatology, 2007, 45( 2): 323- 330. DOI: 10.1002/hep.21460. [8] de KONING TJ, NIKKELS PGJ, DORLAND L, et al. Congenital hepatic fibrosis in 3 siblings with phosphomannose isomerase deficiency[J]. Virchows Arch, 2000, 437( 1): 101- 105. DOI: 10.1007/s004280000185. [9] HENDRIKSZ CJ. Successful treatment of carbohydrate deficient glycoprotein syndrome type 1b with oral mannose[J]. Arch Dis Child, 2001, 85( 4): 339- 340. DOI: 10.1136/adc.85.4.339. [10] HASBAOUI B EL, RIFAI Z, SAGHIR S, et al. Congenital hepatic fibrosis: Case report and review of literature[J]. Pan Afr Med J, 2021, 38: 27941. DOI: 10.11604/pamj.2021.38.188.27941. [11] SRINATH A, SHNEIDER BL. Congenital hepatic fibrosis and autosomal recessive polycystic kidney disease[J]. J Pediatr Gastroenterol Nutr, 2012, 54( 5): 580- 587. DOI: 10.1097/mpg.0b013e31824711b7. [12] XIAO FF, WANG YZ, DONG F, et al. Congenital hepatic fibrosis in a young boy with congenital hypothyroidism: A case report[J]. World J Clin Cases, 2021, 9( 6): 1475- 1482. DOI: 10.12998/wjcc.v9.i6.1475. [13] LEUNG TM, FUNG ML, LIONG EC, et al. Role of nitric oxide in the regulation of fibrogenic factors in experimental liver fibrosis in mice[J]. Histol Histopathol, 2011, 26( 2): 201- 211. DOI: 10.14670/HH-26.201. [14] LEMOINNE S, CADORET A, RAUTOU PE, et al. Portal myofibroblasts promote vascular remodeling underlying cirrhosis formation through the release of microparticles[J]. Hepatology, 2015, 61( 3): 1041- 1055. DOI: 10.1002/hep.27318. [15] DAI WM, LU LG, CAI XB. Relationship between hepatic sinusoidal endothelial cells and hepatic fibrosis[J]. J Clin Hepatol, 2023, 39( 2): 419- 423. DOI: 10.3969/j.issn.1001-5256.2023.02.027.戴伟明, 陆伦根, 蔡晓波. 肝窦内皮细胞与肝纤维化的关系[J]. 临床肝胆病杂志, 2023, 39( 2): 419- 423. DOI: 10.3969/j.issn.1001-5256.2023.02.027. [16] TANG RJ, LIU ZW, SU HB, et al. The clinical features analysis of 48 cases of congenital hepatic fibrosis[J]. Chin Hepatol, 2013, 18( 2): 75- 76. DOI: 10.14000/j.cnki.issn.1008-1704.2013.02.011.汤汝佳, 刘振文, 苏海滨, 等. 先天性肝纤维化48例临床特点分析[J]. 肝脏, 2013, 18( 2): 75- 76. DOI: 10.14000/j.cnki.issn.1008-1704.2013.02.011. -
 本文二维码
本文二维码
计量
- 文章访问数: 403
- HTML全文浏览量: 173
- PDF下载量: 100
- 被引次数: 0

