
 PDF下载 ( 2621 KB)
PDF下载 ( 2621 KB)

进行性家族性肝内胆汁淤积症3型临床病理特征分析
DOI: 10.3969/j.issn.1001-5256.2022.01.024
利益冲突声明:本研究不存在研究者、伦理委员会成员、受试者监护人以及与公开研究成果有关的利益冲突。
作者贡献声明:翁宇航负责收集数据,资料分析,撰写论文;熊清芳、刘杜先、张胥磊指导撰写文章;杨永峰负责课题设计,拟定写作思路,修改论文并最后定稿。
Clinical and pathological features of progressive familial intrahepatic cholestasis type 3
-
摘要:
目的 探讨进行性家族性肝内胆汁淤积症3型(PFIC3)患者临床与病理学特征。 方法 回顾分析了2017年1月—2019年12月南京市第二医院就诊的1326例不明原因肝病患者临床资料,通过临床、病理表现及基因测序确诊PFIC3患者8例(其中1例因禁忌证未行肝组织穿刺)。分析患者临床、检验、影像、病理结果,并对ABCB4相关疾病的病理文献进行回顾,总结PFIC3临床及病理特征。 结果 8例PFIC3患者,其中男5例,女3例,中位年龄29.5岁。50%(4/8)表现为慢性胆汁淤积,50%(4/8)表现胆汁性肝硬化,肝硬化中75%(3/4)合并门静脉高压表现。生化检查中,75%(6/8)表现为ALP升高,100%(8/8)表现GGT升高。影像检查中,50%(4/8)表现为胆囊炎,25%(2/8)表现为胆囊结石,25%(2/8)患者胆管扩张,75%(6/8)患者脾脏肿大,25%(2/8)表现为肝硬化。肝穿刺病理中,所有患者均表现为胆管损伤和/或胆管减少,其中57.1%(4/7)表现为胆管缺失。多耐药蛋白3(MDR3)免疫组化染色42.9%(3/7)正常表达,57.1%(4/7)表达减少。根据文献回顾,其中包含胆管描述或MDR3免疫组化的文献17篇。7例低磷脂相关性胆石症中,胆管正常占71.4%(5/7),胆管减少占14.3%(1/7),胆管缺失占14.3%(1/7);6例妊娠期肝内胆汁淤积症中,胆管正常占16.7%(1/6),胆管减少占50%(3/6),胆管缺失占33.3%(2/6);8例PFIC3中,胆管减少占25%(2/8),胆管缺失占75%(6/8);21例PFIC3患者MDR3表达正常占9.5%(2/21),表达减少占23.8%(5/21),表达缺失占66.7%(14/21)。 结论 PFIC3以胆汁淤积、胆石症、肝纤维化为主要表现。病理表现为胆管损伤,严重者可伴胆管减少或缺失,且损伤程度与疾病严重程度相关。MDR3免疫组化可表现为正常、减少或表达缺失,正常表达患者仍不能排除诊断,必要时行基因检测确诊。 -
关键词:
- 胆汁淤积, 肝内 /
- P糖蛋白类 /
- 病理状态, 体征和症状
Abstract:Objective To investigate the clinical and pathological features of progressive familial intrahepatic cholestasis type 3 (PFIC3). Methods A retrospective analysis was performed for 1326 patients with unexplained liver disease who attended Nanjing Second Hospital from January 2017 to December 2019, among whom 8 patients were diagnosed with PFIC3 based on clinical/pathological manifestation and gene sequencing results (1 patient did not undergo liver biopsy due to contraindication). Clinical, laboratory, imaging, and pathological findings were analyzed and a literature review was performed for the pathology of ABCB4-related diseases to summarize the clinical and pathological features of PFIC-3. Results Among the 8 patients with PFIC3, there were 5 male patients and 3 female patients, with a median age of 29.5 years. Of all 8 patients, 4 (50%) manifested as chronic cholestasis and 4 (50%) manifested as biliary cirrhosis, among whom 3 (75%) had the manifestation of portal hypertension. As for biochemical examination, 75% (6/8) had an increase in alkaline phosphatase, and 100% (8/8) had an increase in gamma-glutamyl transpeptidase. As for imaging examination, 50% (4/8) had cholecystitis, 25% (2/8) had gallstones, 25% (2/8) had bile duct dilatation, 75% (6/8) had splenomegaly, and 25% (2/8) had liver cirrhosis. As for liver biopsy, all 7 patients manifested as bile duct injury and/or reduction, and 57.1% (5/7) had absence of the bile duct. Multidrug resistance P-glycoprotein 3 (MDR3) immunohistochemical staining showed normal expression in 42.9% (3/7) of the patients and reduced expression in 57.1% (4/7) of the patients. Literature review obtained 17 articles with a description of the bile duct or MDR3 immunohistochemistry. Among the 7 patients with low phospholipid-associated cholelithiasis, 71.4% (5/7) had normal bile duct, 14.3% (1/7) had bile duct reduction, and 14.3% (1/7) had absence of the bile duct; among the 6 patients with intrahepatic cholestasis of pregnancy, 16.7% (1/6) had normal bile duct, 50% (3/6) had bile duct reduction, and 33.3% (2/6) had absence of the bile duct; among the 8 patients with PFIC3, 25% (2/8) had bile duct reduction and 75% (6/8) had absence of bile duct; among the 21 patients with PFIC3, 9.5% (2/21) had normal expression of MDR3, 23.8% (5/21) had a reduction in the expression of MDR3, and 66.7% (14/21) had absence of the expression of MDR3. Conclusion PFIC3 mainly manifests as cholestasis, cholelithiasis, and hepatic fibrosis. Pathological manifestation includes bile duct injury and bile duct reduction or absence of the bile duct in severe cases, and the degree of injury is associated with disease severity. MDR3 immunohistochemistry may show normal expression, reduced expression, or absence of expression, and diagnosis cannot be excluded in patients with normal expression. Genetic testing can be performed for diagnosis when necessary. -
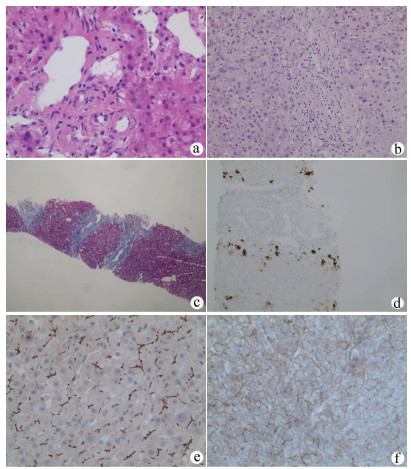
图 1 PFIC3患者肝组织病理表现
注:a,病例2汇管区小叶间胆管可见上皮细胞缺失,胆管损伤(HE染色,×400);b,病例4汇管区纤维增生,未见和小叶间动脉伴行的小叶间胆管,胆管缺失(HE染色,×200);c,病例1以汇管区为中心的纤维增生形成假小叶(Masson染色,×100);d,病例7显示一个较大的汇管区胆管缺失,界面处可见细胆管,部分肝细胞CK7表达(免疫组化,×200);e,病例2 MDR3沿肝细胞毛细胆管面表达,表达量基本正常(免疫组化,×400);f,病例6约20%肝细胞毛细胆管面MDR3表达,表达明显减少,部分肝细胞浆MDR3弱表达(免疫组化,×400)。
表 1 8例PFIC3患者临床特征
编号 性别 年龄 临床表现 影像表现 ABCB4突变类型 突变意义 1 男 25 慢性胆汁淤积、肝硬化、门静脉高压、上消化道出血 肝硬化、胆囊炎、胆囊结石、脾肿大 复合杂合 软件分析可能致病 2 男 21 慢性胆汁淤积 肝右叶钙化灶、胆囊炎、脾轻度肿大 复合杂合 国内外文献均报道,软件分析可能致病 3 女 54 慢性胆汁淤积 胆囊炎、胆囊结石、脾肿大、脾脏低密度结节 杂合 软件分析可能致病 4 男 18 慢性胆汁淤积 肝内胆管扩张 杂合 软件分析可能致病 5 男 31 慢性胆汁淤积、肝硬化 肝右叶囊肿、胆囊壁毛糙、脾脏肿大 杂合 软件分析可能致病 6 女 38 慢性胆汁淤积、肝硬化、门静脉高压 肝脏形态失常、肝右叶胆管轻度扩张 复合杂合 文献报道可能影响剪切 7 男 37 慢性胆汁淤积、肝硬化、门静脉高压、上消化道出血 肝硬化表现、脾肿大、门静脉增粗、门静脉高压、胆囊炎 杂合 软件分析可能影响剪切 8 女 28 慢性胆汁淤积 肝内钙化灶、胆囊壁毛糙、脾轻度肿大 杂合 软件分析可能致病  下载: 导出CSV
下载: 导出CSV
表 2 8例PFIC3患者实验室检查
指标 病例1 病例2 病例3 病例4 病例5 病例6 病例7 病例8 RBC(×109/L) 2.74~4.23 4.52~4.77 3.69~4.06 4.01~4.84 4.27 2.81~3.01 4.07~4.27 4.42 Hb(g/L) 79~108 132~138 116~125 120~146 138 82~84 116~119 143 WBC(×109/L) 1.57~3.13 4.85~6.51 2.34~2.37 2.78~5.34 8.24 12.72~15.91 2.19~2.39 5.49 PLT(×109/L) 23~54 137~189 55~65 91~158 159 367~403 42~65 278 INR 1.23~1.25 0.91~0.99 1.06 0.97 1.14 1.14~1.20 1.10 0.96 TBil(μmol/L) 84.70~195.30 15.80~33.40 18.10~27.60 18.60~25.90 105 72.40~108.50 19.90~25.50 9.00~15.90 ALT(U/L) 98.3~420.1 42.9~237.1 31.6~71.0 76.0~126.6 95.7 50.0~57.4 51.5~128.3 29.4~126.1 AST(U/L) 93.1~632.6 58.2~186.7 31.8~55.7 34.4~53.5 42.9 140.5~150.0 44.0~106.4 18.2~74.4 Alb(g/L) 30.8~39.9 41.2~45.2 36.1~37.7 41.2~54.2 53 20.6~21.0 38.2~39.0 40.2~45.2 ALP(U/L) 554.5~1 202.4 372.1~989.5 177.1~206.5 91~223 88.9 467.6~528.8 386.6~514.2 51~65 GGT(U/L) 364.9~575.6 769.7~1 644.9 263.3~389.1 205.8~321.1 86.8 161.1~232.6 219.4~348.9 86~157 LDH(U/L) 129~251 185~226 171~194 125~147 177 281~322 143~163 143~162
下载: 导出CSV
表 3 PFIC3患者组织病理学及免疫组化染色特征
病例 胆管损伤程度 炎症分级(G)、纤维分期(S) 肝细胞CK7表达 MDR3表达 1 胆管缺失 G1、S4 有 正常 2 胆管减少 G2、S2 无 正常 3 胆管减少 G2、S2 无 减少 4 胆管缺失 G1、S3 有 减少 6 胆管缺失 G2、S4 有 减少 7 胆管缺失 G1、S3~4 偶见 减少 8 胆管减少 G1、S2 无 正常
下载: 导出CSV
-
[1] BULL LN, THOMPSON RJ. Progressive familial intrahepatic cholestasis[J]. Clin Liver Dis, 2018, 22(4): 657-669. DOI: 10.1016/j.cld.2018.06.003. [2] GUNAYDIN M, BOZKURTER CIL AT. Progressive familial intrahepatic cholestasis: Diagnosis, management, and treatment[J]. Hepat Med, 2018, 10: 95-104. DOI: 10.2147/HMER.S137209. [3] REICHERT MC, LAMMERT F. ABCB4 gene aberrations in human liver disease: An evolving spectrum[J]. Semin Liver Dis, 2018, 38(4): 299-307. DOI: 10.1055/s-0038-1667299. [4] YANG YF. Road map of diagnosis in patients with unknown causes[J]. J Prac Hepatol, 2018, 21(1): 1-3. DOI: 10.3969/j.issn.1672-5069.2018.01.001.杨永峰. 不明原因肝病诊断思路[J]. 实用肝脏病杂志, 2018, 21(1): 1-3. DOI: 10.3969/j.issn.1672-5069.2018.01.001. [5] LIPIŃSKI P, CIARA E, JURKIEWICZ D, et al. Targeted next-generation sequencing in diagnostic approach to monogenic cholestatic liver disorders-single-center experience[J]. Front Pediatr, 2020, 8: 414. DOI: 10.3389/fped.2020.00414. [6] DENG M, GUO L, SONG YZ. Clinical and genetic analysis of a family affected by progressive familial intraphepatic cholestasis type 3[J]. Chin J Med Genetics, 2018, 35(5): 686-690. DOI: 10.3760/cma.j.issn.1003-9406.2018.05.015.邓梅, 郭丽, 宋元宗. 一例进行性家族性肝内胆汁淤积症3型患儿的临床及遗传学分析[J]. 中华医学遗传学杂志, 2018, 35 (5): 686-690. DOI: 10.3760/cma.j.issn.1003-9406.2018.05.015. [7] Chinese Society of Hepatology, Chinese Medical Association; Chinese Society of Gastroenterology, Chinese Medical Association; Chinese Society of Infectious Diseases, Chinese Medical Association. Consensus on the diagnosis and treatment of cholestasis liver diseasess[J]. J Clin Hepatol, 2015, 31(12): 1989-1999. DOI: 10.3760/cma.j.issn.1007-3418.2015.12.004.中华医学会肝病学分会, 中华医学会消化病学分会, 中华医学会感染病学分会. 胆汁淤积性肝病诊断和治疗共识(2015)[J]. 临床肝胆病杂志, 2015, 31(12): 1989-1999. DOI: 10.3760/cma.j.issn.1007-3418.2015.12.004. [8] Chinese Society of Hepatology, Chinese Medical Association. Chinese guidelines on the management of liver cirrhosis[J]. J Clin Hepatol, 2019, 35(11): 2408-2425. DOI: 10.3760/cma.j.issn.1007-3418.2019.11.008.中华医学会肝病学分会. 肝硬化诊治指南[J]. 临床肝胆病杂志, 2019, 35(11): 2408-2425. DOI: 10.3760/cma.j.issn.1007-3418.2019.11.008. [9] JACQUEMIN E. Progressive familial intrahepatic cholestasis[J]. Clin Res Hepatol Gastroenterol, 2012, 36(Suppl 1): s26-s35. DOI: 10.1016/S2210-7401(12)70018-9. [10] TIAN AP, YANG YF. A comparative analysis of pathological grading and staging systems for chronic hepatitis[J]. J Clin Hepatol, 2018, 34(11): 2271-2277. DOI: 10.3969/j.issn.1001-5256.2018.11.002.田爱平, 杨永峰. 慢性肝炎病理学分级分期评分系统比较[J]. 临床肝胆病杂志, 2018, 34(11): 2271-2277. DOI: 10.3969/j.issn.1001-5256.2018.11.002. [11] YANG YF. Atlas of liver pathology[M]. Changsha: Central South University Publishing Group, 2018: 19-40.杨永峰主译. 肝脏病理学图解[M]. 长沙: 中南大学出版社, 2018: 19-40. [12] YAO GB. Clinical hepatology[M]. Shanghai: Shanghai Scientific & Technical Publishers, 2011: 344-345.姚光弼. 临床肝脏病学[M]. 上海: 上海科学技术出版社, 2011: 344-345. [13] FUSSEY SP, GUEST JR, JAMES OF, et al. Identification and analysis of the major M2 autoantigens in primary biliary cirrhosis[J]. Proc Natl Acad Sci U S A, 1988, 85(22): 8654-8658. DOI: 10.1073/pnas.85.22.8654. [14] XIANG D, HE J, WANG H, et al. Liver transplantation for decompensated liver cirrhosis caused by progressive familial intrahepatic cholestasis type 3: A case report[J]. Medicine (Baltimore), 2017, 96(50): e9158. DOI: 10.1097/MD.0000000000009158. [15] GOTTHARDT D, RUNZ H, KEITEL V, et al. A mutation in the canalicular phospholipid transporter gene, ABCB4, is associated with cholestasis, ductopenia, and cirrhosis in adults[J]. Hepatology, 2008, 48(4): 1157-1166. DOI: 10.1002/hep.22485. [16] KHABOU B, SIALA-SAHNOUN O, GARGOURI L, et al. In silico investigation of the impact of synonymous variants in ABCB4 gene on mRNA stability/structure, splicing accuracy and codon usage: Potential contribution to PFIC3 disease[J]. Comput Biol Chem, 2016, 65: 103-109. DOI: 10.1016/j.compbiolchem.2016.10.008. [17] GOUBRAN M, ADERIBIGBE A, JACQUEMIN E, et al. Case report: Progressive familial intrahepatic cholestasis type 3 with compound heterozygous ABCB4 variants diagnosed 15 years after liver transplantation[J]. BMC Med Genet, 2020, 21(1): 238. DOI: 10.1186/s12881-020-01173-0. [18] WU Z, ZHANG S, ZHANG L, et al. Novel ABCB4 mutation in a Chinese female patient with progressive familial intrahepatic cholestasis type 3: A case report[J]. Diagn Pathol, 2020, 15(1): 39. DOI: 10.1186/s13000-020-00955-7. [19] COLOMBO C, VAJRO P, DEGIORGIO D, et al. Clinical features and genotype-phenotype correlations in children with progressive familial intrahepatic cholestasis type 3 related to ABCB4 mutations[J]. J Pediatr Gastroenterol Nutr, 2011, 52(1): 73-83. DOI: 10.1097/MPG.0b013e3181f50363. [20] de VRIES E, MAZZETTI M, TAKKENBERG B, et al. Carriers of ABCB4 gene variants show a mild clinical course, but impaired quality of life and limited risk for cholangiocarcinoma[J]. Liver Int, 2020, 40(12): 3042-3050. DOI: 10.1111/liv.14662. [21] DZAGANIA T, ENGELMANN G, HÄUSSINGER D, et al. The histidine-loop is essential for transport activity of human MDR3. A novel mutation of MDR3 in a patient with progressive familial intrahepatic cholestasis type 3[J]. Gene, 2012, 506(1): 141-145. DOI: 10.1016/j.gene.2012.06.029. [22] STICOVA E, NEROLDOVA M, KOTALOVA R, et al. ABCB4 disease mimicking morbus Wilson: A potential diagnostic pitfall[J]. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub, 2020, 164(1): 121-125. DOI: 10.5507/bp.2019.054. [23] BOGA S, JAIN D, SCHILSKY ML. Presentation of progressive familial intrahepatic cholestasis type 3 mimicking wilson disease: Molecular genetic diagnosis and response to treatment[J]. Pediatr Gastroenterol Hepatol Nutr, 2015, 18(3): 202-208. DOI: 10.5223/pghn.2015.18.3.202. [24] GIOVANNONI I, SANTORELLI FM, CANDUSSO M, et al. Two novel mutations in African and Asian children with progressive familial intrahepatic cholestasis type 3[J]. Dig Liver Dis, 2011, 43(7): 567-570. DOI: 10.1016/j.dld.2011.03.004. [25] de VREE JM, JACQUEMIN E, STURM E, et al. Mutations in the MDR3 gene cause progressive familial intrahepatic cholestasis[J]. Proc Natl Acad Sci U S A, 1998, 95(1): 282-287. DOI: 10.1073/pnas.95.1.282. [26] WENDUM D, BARBU V, ROSMORDUC O, et al. Aspects of liver pathology in adult patients with MDR3/ABCB4 gene mutations[J]. Virchows Arch, 2012, 460(3): 291-298. DOI: 10.1007/s00428-012-1202-6. [27] FANG LJ, WANG XH, KNISELY AS, et al. Chinese children with chronic intrahepatic cholestasis and high γ-glutamyl transpeptidase: Clinical features and association with ABCB4 mutations[J]. J Pediatr Gastroenterol Nutr, 2012, 55(2): 150-156. DOI: 10.1097/MPG.0b013e31824ef36f. [28] ZIOL M, BARBU V, ROSMORDUC O, et al. ABCB4 heterozygous gene mutations associated with fibrosing cholestatic liver disease in adults[J]. Gastroenterology, 2008, 135(1): 131-141. DOI: 10.1053/j.gastro.2008.03.044. [29] VIJ M, VALAMPARAMPIL J, SHANMUGUM N, et al. Paucity of interlobular bile ducts in Multidrug-Resistant P-Glycoprotein 3 (MDR3) deficiency[J]. Int J Surg Pathol, 2019, 27(3): 343-347. DOI: 10.1177/1066896918799941. [30] FRIDER B, CASTILLO A, GORDO-GILART R, et al. Reversal of advanced fibrosis after long-term ursodeoxycholic acid therapy in a patient with residual expression of MDR3[J]. Ann Hepatol, 2015, 14(5): 745-751. [31] SANNIER A, GANNE N, TEPPER M, et al. MDR3 immunostaining on frozen liver biopsy samples is not a sensitive diagnostic tool for the detection of heterozygous MDR3/ABCB4 gene mutations[J]. Virchows Arch, 2012, 460(5): 535-537; author reply 539. DOI: 10.1007/s00428-012-1231-1. [32] GOTTHARDT D, RUNZ H, KEITEL V, et al. A mutation in the canalicular phospholipid transporter gene, ABCB4, is associated with cholestasis, ductopenia, and cirrhosis in adults[J]. Hepatology, 2008, 48(4): 1157-1166. DOI: 10.1002/hep.22485. [33] FOMBUENA B, AMPUERO J, ÁLVAREZ L, et al. LPAC syndrome associated with deletion of the full exon 4 in a ABCB4 genetic mutation in a patient with hepatitis C[J]. Rev Esp Enferm Dig, 2014, 106(8): 544-547. [34] ROSMORDUC O, HERMELIN B, BOELLE PY, et al. ABCB4 gene mutations and primary sclerosing cholangitis[J]. Gastroenterology, 2004, 126(4): 1220-1222; author reply 1222-1223. DOI: 10.1053/j.gastro.2004.02.042. [35] DAVIT-SPRAUL A, GONZALES E, BAUSSAN C, et al. The spectrum of liver diseases related to ABCB4 gene mutations: Pathophysiology and clinical aspects[J]. Semin Liver Dis, 2010, 30(2): 134-146. DOI: 10.1055/s-0030-1253223. [36] MOROTTI RA, SUCHY FJ, MAGID MS. Progressive familial intrahepatic cholestasis (PFIC) type 1, 2, and 3: A review of the liver pathology findings[J]. Semin Liver Dis, 2011, 31(1): 3-10. DOI: 10.1055/s-0031-1272831. [37] JACQUEMIN E, de VREE JM, CRESTEIL D, et al. The wide spectrum of multidrug resistance 3 deficiency: From neonatal cholestasis to cirrhosis of adulthood[J]. Gastroenterology, 2001, 120(6): 1448-1458. DOI: 10.1053/gast.2001.23984. [38] THOENI C, WALDHERR R, SCHEUERER J, et al. Expression analysis of ATP-binding cassette transporters ABCB11 and ABCB4 in primary sclerosing cholangitis and variety of pediatric and adult cholestatic and noncholestatic liver diseases[J]. Can J Gastroenterol Hepatol, 2019, 2019: 1085717. DOI: 10.1155/2019/1085717. [39] de VRIES E, MAZZETTI M, TAKKENBERG B, et al. Carriers of ABCB4 gene variants show a mild clinical course, but impaired quality of life and limited risk for cholangiocarcinoma[J]. Liver Int, 2020, 40(12): 3042-3050. DOI: 10.1111/liv.14662. [40] STICOVA E, JIRSA M. ABCB4 disease: Many faces of one gene deficiency[J]. Ann Hepatol, 2020, 19(2): 126-133. DOI: 10.1016/j.aohep.2019.09.010. [41] STÄTTERMAYER AF, HALILBASIC E, WRBA F, et al. Variants in ABCB4 (MDR3) across the spectrum of cholestatic liver diseases in adults[J]. J Hepatol, 2020, 73(3): 651-663. DOI: 10.1016/j.jhep.2020.04.036. [42] COLOMBO C, VAJRO P, DEGIORGIO D, et al. Clinical features and genotype-phenotype correlations in children with progressive familial intrahepatic cholestasis type 3 related to ABCB4 mutations[J]. J Pediatr Gastroenterol Nutr, 2011, 52(1): 73-83. DOI: 10.1097/MPG.0b013e3181f50363. -
 本文二维码
本文二维码
图(1) / 表(4)
计量
- 文章访问数: 1012
- HTML全文浏览量: 365
- PDF下载量: 104
- 被引次数: 0

